单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
Cell Ranger是一个“傻瓜”软件,你只需提供原始的fastq文件,它就会返回feature-barcode表达矩阵。为啥不说是gene-cell,举个例子,cell hashing数据得到的矩阵还有tag行,而列也不能肯定就是一个cell,可能考虑到这个才不叫gene-cell矩阵吧~它是10xgenomics提供的官方比对定量软件,有四个子命令,我只用过cellranger count,另外三个cellranger mkfastq、cellranger aggr、cellranger reanalyze没用过,也没啥影响。
下载:https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
安装:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation
在讲Cell Ranger的使用之前,先来看一下10X的单细胞数据长什么样
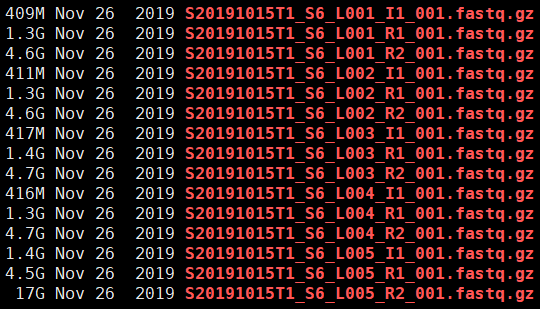
这是一个样本5个Line的测序数据,数据量足够的话可能只有一个Line。可以看出,它们的命名格式相对规范,在收到公司的数据后,尽量不要自己更改命名。此外还要注意一个细节,就是存放这些fastq文件的目录应该用第一个下划线
_前面的字符串命名,否则后续cell ranger将无法识别目录里面的文件,同时报错
[error] Unable to detect the chemistry for the following dataset. Please validate it and/or specify the chemistry via the --chemistry argument.
其实并不是--chemistry参数的问题。
为了更清楚地理解文件内容,我们来看一下10X单细胞的测序示意图

Read1那一段序列原本是连在磁珠上面的,有cellular barcode(一个磁珠上都一样),有UMI(各不相同),还有poly-T。Read2就是来源于细胞内的RNA。它俩连上互补配对之后,还会在Read2的另一端连上sample index序列。这段sample index序列的作用是什么呢?可以参考illumina测序中index primers的作用:

简单来说就是为了在一次测序中,测多个样本,在来源于特定样本的序列后都加上特定的index,测完之后根据对应关系拆分。一个样本对应4个index:

再看每个文件里面是什么就容易理解了,我们以一个Line为例:
less -S S20191015T1_S6_L001_I1_001.fastq.gz | head -n 8 less -S S20191015T1_S6_L001_R1_001.fastq.gz | head -n 8 less -S S20191015T1_S6_L001_R2_001.fastq.gz | head -n 8
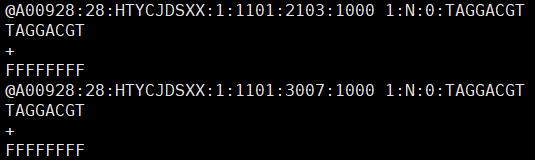
其实这个index序列就包含在文件的第1、5、9...行,有点多余,一般不太关注它。这个文件的序列最多四种,感兴趣的小伙伴可以看看。

R1文件里面就是cellular barcode信息,多余的序列已经去掉了。10X的v2试剂碱基长度是26,v3试剂碱基长度是28

最后一个文件就是真正的转录本对应的cDNA序列
上一篇讲到cell hashing测序有转录本信息,得到的文件和上面是一样的;还有一个细胞表面蛋白信息,根据这个蛋白信息区分细胞来源,如下:

从图中可以看出,和普通转录本建库差不多,就是R2那一部分换成了HTO序列,整个片段长度也改变了。


上面两张图是我在实际处理中看到的两种cell hashing测序,第一张是TotalSeqA,第二张是TotalSeqB。TotalSeqA中,R2第一个碱基开始为HTO序列(之后是polyA序列),而TotalSeqB中,R2前10个碱基为N的任意碱基,第11个碱基为HTO序列的开始位置,HTO序列长度为16。
综上,cell hashing的测序数据有两套,一套是常规的转录本fastq,一套是蛋白信息(也可以说是样本信息)的fastq。所以处理这类数据,要跟测序公司确认清楚用的是TotalSeqA还是B,以及样本和HTO序列的对应关系。
接
56c
下来说说如何用Cell Ranger处理普通10X单细胞测序数据,以及cell hashing单细胞测序数据
普通10X
indir=/project_2019_11/data/S20191015T1
outdir=/project_2019_11/cellranger/
sample=S20191015T1
ncells=5000 #预计细胞数,这个参数对最终能得到的细胞数影响并不大,所以不用纠结
threads=20
refpath=/ref/10x/human/refdata-cellranger-GRCh38-3.0.0
cellranger=/softwore/bin/cellranger
cd ${outdir}
${cellranger} count --id=${sample} \
--transcriptome=${refpath} \
--fastqs=${indir} \
--sample=${sample} \
--expect-cells=${ncells} \
--localcores=${threads}
cell hashing
total_seq_A
需要提前准备好两个文件夹,比如我用total_seq_A或total_seq_B存放HTO序列和样本来源的对应关系:
$ ls feature.reference1.csv $ cat feature.reference1.csv id,name,read,pattern,sequence,feature_type tag1,tag1,R2,^(BC),GTCAACTCTTTAGCG,Antibody Capture tag2,tag2,R2,^(BC),TGATGGCCTATTGGG,Antibody Capture
tag1、tag2对应哪一个样本事先知道;^(BC)可以看做正则表达式,表示R2序列以barcode(也就是HTO序列)开始
total_seq_B
$ ls feature.reference.csv $ cat fe 56c ature.reference.csv id,name,read,pattern,sequence,feature_type tag6,tag6,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,GGTTGCCAGATGTCA,Antibody Capture tag7,tag7,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,TGTCTTTCCTGCCAG,Antibody Capture
5PNNNNNNNNNN(BC)NNNNNNNNN表示从5端开始,10个碱基之后就是HTO序列,后面的序列随意
lib_csv
第二个文件夹lib_csv,用来存放cell hashing两套数据的路径,用csv格式存储,sample这一列为文件夹名称
$ cat S20200612P1320200702N.libraries.csv fastqs,sample,library_type /project_2019_11/data/fastq/,S20200612P1320200702N,Gene Expression /project_2019_11/data/antibody_barcode/,S20200612P13F20200702N,Antibody Capture
最终脚本如下
lib_dir=/script/cellranger/1/lib_csv/
#need to be changed based on your seq-tech: total_seq_A or total_seq_B
feature_ref_dir=/script/cellranger/1/total_seq_A/
outdir=/project_2019_11/cellranger/
sample=S20191017P11
ncells=5000
threads=20
refpath=/ref/10x/human/refdata-cellranger-GRCh38-3.0.0
cellranger=/softwore/bin/cellranger
cd ${outdir}
${cellranger} count --libraries=${lib_dir}${sample}.libraries.csv \
--r1-length=28 \
--feature-ref=${feature_ref_dir}feature.reference1.csv \
--transcriptome=${refpath} \
--localcores=${threads} \
--expect-cells=${ncells} \
ad8
--id=${sample}
最终的表达矩阵会输出到
${outdir}${sample_id}/outs/filtered_feature_bc_matrix
$ cd S20200619P11120200716NC/outs/filtered_feature_bc_matrix/ $ ls barcodes.tsv.gz features.tsv.gz matrix.mtx.gz $ less -S features.tsv.gz ENSG00000243485 MIR1302-2HG Gene Expression ENSG00000237613 FAM138A Gene Expression ...... ENSG00000277475 AC213203.1 Gene Expression ENSG00000268674 FAM231C Gene Expression tag7 tag7 Antibody Capture tag8 tag8 Antibody Capture
features.tsv.gz存储的是基因信息,因为是cell hashing数据,矩阵最后多了几行tag信息,共33540行
$ less -S barcodes.tsv.gz | head -n 4 AAACCCAAGACTTAAG-1 AAACCCAAGCTACTGT-1 AAACCCAAGGACTGGT-1 AAACCCAAGGCCTGCT-1
barcodes.tsv.gz存放的是最后得到的cellular barcode,共10139行
$ less -S matrix.mtx.gz | head -n 8
%%MatrixMarket matrix coordinate integer general
%metadata_json: {"format_version": 2, "software_version": "3.1.0"}
33540 10139 15746600
65 1 1
103 1 1
155 1 2
179 1 2
191 1 1
matrix.mtx.gz为矩阵信息,除前三行外,余下的行数等于feature乘以CB数,第二列表示CB编号,从1到10139,1重复33540次,对应第一列的33540个feature。第三列表示UMI
下面的脚本可以将这三个文件转换为常见的矩阵形式
path1=/softwore/biosoft/cellranger-3.1.0/cellranger
path2=/project_2019_11/cellranger/
i=S20191211P71
${path1} mat2csv ${path2}${i}/outs/filtered_feature_bc_matrix ${path2}Feature_Barcode_Matrices/${i}.mat.count.csv
sed 's/,/\t/g' ${path2}Feature_Barcode_Matrices/${i}.mat.count.csv > ${path2}Feature_Barcode_Matrices/${i}.mat.count.txt
sed -i 's/^\t//g' ${path2}Feature_Barcode_Matrices/${i}.mat.count.txt
rm -f ${path2}Feature_Barcode_Matrices/${i}.mat.count.csv

- 单细胞测序数据的差异表达分析方法总结-single cell differential expression
- 单细胞分析实录(3): Cell Hashing数据拆分
- 使用ICP得到的变换矩阵更新相机位置
- ArcGIS 网络分析[2] 在ArcMap中使用网络数据集进行五大网络分析[最短路径/服务区/最近设施点/OD成本矩阵/车辆分配]
- RACI 职责分配矩阵 模型使用详解及案例分析
- 单细胞文献分析 Quantitative single-cell rna-seq with unique molecular identifers
- 单细胞分析实录(8): 展示marker基因的4种图形(一)
- Android 使用Matrix进行图像变换,和矩阵分析!
- 使用关系数据库和矩阵,Reporting Service也能实现类似Analysis的分析
- Matlab 1.矩阵的切割再把得到的矩阵按顺序拼接排列(1维数据按规律转为2维数据方便深度学习使用)
- 单细胞分析实录(6): 去除批次效应/整合数据
- 单细胞分析实录(5): Seurat标准流程
- 使用clementine得到混淆矩阵
- 使用AutoHotkey分析日志文件,得到分析报告。
- Scull字符设备驱动模块的内存使用分析
- Skyeye使用经验及分析一例.
- Rational Purify 使用及分析实例
- MFC中数组类的使用分析
- Oracle 分析函数的使用
- 综合门户网站矩阵模式分析