三代测序技术概况
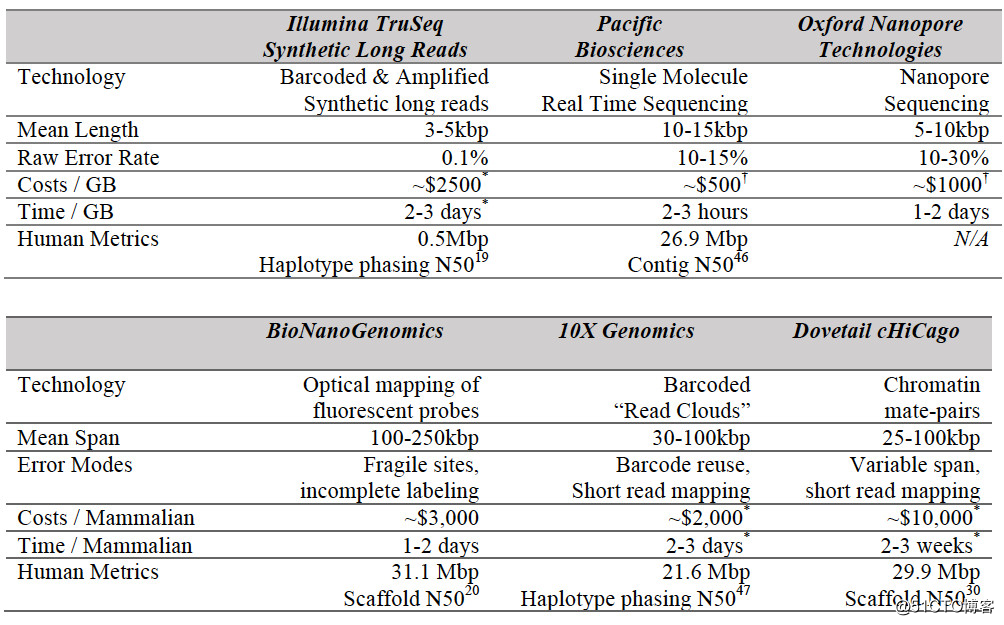
第三代长序列测序技术为获得高质量的基因组数据提供了机遇。二代测序会产生很多数百个碱基大小的读长,而三代测序的读长可以长达10kbp。这种长读长对基因组的从头组装、基因组结构变异和基因分型(phasing)有重要的意义。
自从二代测序技术商业化以来,很多测序平台相继出现,比如Roche/454(2005年),Illumina/Solexa(2007年)等。这些测序平台使得测序价格大幅下降。因此,人们可以测定很多新物种序列,同时还能够研究不同种群的基因组多样性。但是二代测序对研究基因组结构变异却十分困难。而且,通过二代测序技术对新基因组的从头测序结果也并不理想,甚至不如之前老的方法准确,容易造成基因组片段的缺失。即便是对基因组的重测序也很难进行基因组结构变异的研究。
不过通过单分子测序则能很大程度上解决这些二代测序技术的不足。单分子测序的读长能够达到10kbp,甚至超过100kbp。这么大的长度对研究基因组结构变异提供了很大的方便。
更为重要的是,长读长能够很准确的显示出重复序列,或者连续性更好的基因组。还能够很容易的识别出插入缺失突变、转座、倒置等结构变异。同时单分子测序的测序深度在基因组上分布比较均一,不会像二代测序那样受序列内容影响(如GC含量)造成很多区域的测序深度降低甚至缺失。【GC含量高的序列容易造成测序深度偏低】。通过这种第三代长测序技术能够形成一个超级contig(scaffold),有时甚至可以覆盖整个染色体的一条臂。
三代测序技术已经用于了很多微生物基因组的高精度从头组装、动植物基因组的连续性重构。同时也可以用了重测序分析,比如获得人类染色体的结构变异图和分型变异图。尤其是这些新技术的应用,填补了在人类参考基因组中存在的序列间隔。此外,读长的提升在临床上也有很重要应用,比如对人类主要组织兼容复合物(HLA)的测序。在宏基因组中,通过长序列测序,能够解决不同种群个体混杂的问题。三代测序还可以用于转录组的研究以及表观遗传修饰的研究。总之,相比于二代测序,三代技术带来的三大特点(‘3C’):连贯性(contiguity)、完整性(completeness)和准确性(correctness).
目前有三种商用第三代测序平台:PacBio的SingleMolecule Real Time(SMRT)测序、Illumina的Tru-seqSynthetic Long-Read测序,和Oxford Nanopore测序。这些测序平台可以产生5kbp到15kbp的测序片段,有些可达100kbp。
当然其中最成熟的还是PacBio的SMRT,其在2010年开始商用。SMRT也是使用边合成边测序的技术,通过荧光标记的碱基来识别DNA序列。比如PacBio RSII测序平台,能够测得100kbp读长,每天产生8GB的数据量。原始测序错误在10%-15%,不过通过公式校正可以将每个碱基的准确率提升到99.99%。不过PacBio的不足时价格比较高,这也限制了其大规模的使用。尽管如此,还是有不少研究通过PacBio对微生物、真菌、动植物的基因组进行了测序和组装,也包括人的。
第二个三代测序技术是在2012年由Illumina发明的TruSeq Synthetic Long Reads,是通过短读长序列得来的,所以其准确度非常高,错误率只有0.1%,因而可以不用校正直接用来基因分型分析和组装。它的缺点是读长相对于其他三代测序要短一些,而且容易受到GC偏倚影响。此外,如果是从头组装基因组,那么对短读长的测序深度可能会达到900X到1500X,这样最后才能获得30X的长读长序列。
最新的三代测序技术在2014年,来自Oxford Nanopore。其最新的测序平台MinION非常小,携带很方便。它的测序读长和PacBio相似。不过它的准确度很低,测序通量也不高,因而它的使用目前主要针对基因组较小的生物,比如大肠杆菌和酵母菌。通过校正,每个碱基的准确性也能提升到99.95%。但是由于其体积非常小,花费很低,非常适合在偏远的地方使用,比如在西非爆发埃博拉的地区。

(本人实拍,Nanodrop的MinION测序仪器)
第三代基因图谱
基因图谱能够帮助我们在不了解每一个碱基序列的情况下知道DNA的序列结构。可以通过分析杂合子标记之间的重组率来重建基因图谱。但这需要很大的样本量,对于某些物种来说是很难实现的。二代基因图谱使用了配对文库建立。最成功的第三代基因图谱是在2010年来自BioNano Genomics 的Irys。通过PacBio测序和Irys基因图谱完成了至今连贯性最好的从头组装的人类基因组,contig的N50达到了1.4Mbp,同时在基因组中发现了数百的新的结构变异。在2015年初,Dovetail Genomics通过优化Hi-C的方法发明了cHiCago方法,这种方法使得基因图谱的构建相对便宜,不过这个方法属于Dovetail专有,样本必须寄到他们公司有他们内部完成构建。最新的基因图谱构建技术来自10XGenomics。它的原理和Illunima的长测序原理相似。
基因组装:基因组装最大的障碍来自基因组中的重复序列。二代测序对重复序列,尤其是比读长而还长的重复序列的组装无能为力。相比之下,三代测序由于其读长很长,在对重复序列的组装中发挥了巨大作用。
长读长的组装使用overlapgraph或者stringgraph来完成。IlluminaTru-seq的准确性很高,因而可以直接用来组装,而PacBio和MinION的准确性低,因为需要在组装之前最数据进行校正。三代测序产生的读长分布通常是对数正态分布。
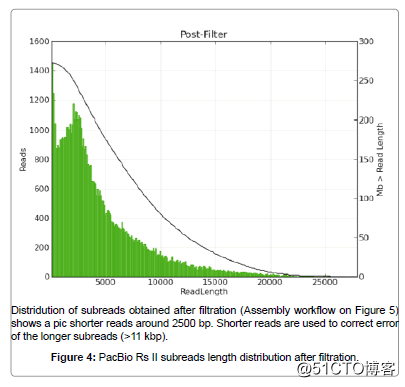
这种分布就意味着,大多数读长是很短的,只有少数读长可能达到100kbp。所以即便是三代测序技术,保证一定的测序深度对基因组的组装依然是十分重要的。
结构变异分析:如果是仅仅研究像SNP一样的很小的变异,二代测序就能够胜任;但是如果要研究很大的结构变异(>50bp),则二代测序的短读长很难识别变异位点。三代测序的长读长能够很有效的识别出结构变异位点。比如通过三代测序技术,在人类基因组中发现了数万个结构变异,而这些变异通常无法通过二代测序识别。
基因分型:即将杂合子个体的变异分配到不同的单倍体上。基因分型会受到测序错误和测序深度偏倚的影响,可能因此引入错误的变异类型或者错失真正的杂合子变异类型。在人类基因组中,杂合子变异在染色体上的距离为1000bp– 1500bp,这个距离显然超过了二代测序的读长,而三代测序则能够很准确的对此进行分型。
第三代测序技术大大提高了基因组的质量,对于大多数基因组<100Mbp的生物,其基因组可以通过第三代测序进行完美的组装;对于更大的基因组,如人类和其他哺乳动物,其基因组的组装质量也有很大幅度提升。
三代测序的三大特性
连贯性:连贯性对基因组的组装非常重要,如果连贯性比较好,能够准确的反应出基因结构之间的关系(外显子、基因簇、转移元件、调节序列等)。早在1988年就有了Lander-Waterman模型来描述基因的连贯性,估计最低测序深度,并且预测了基于不同读长的contig的平均长度。不过这种预测在测序深度很大的条件下很不准确,比如其预测在100bp读长100X的测序深度下,可以组装成数百GB大小的contig,显然这已经超多了人类基因组本身的大小。
Lander-Waterman预测不准确的一个原因是其忽略了基因组中的重复序列。重复序列的大小分布是按照指数形式递减的,也就是绝大多数重复序列都是很短的,所以哪怕是测序读长稍微增加一些,就能解决掉很大一部分重新序列的组装问题。
完整性:如果一个基因组的测序深度>50X,理论上每一个碱基都会被测到。但实际上,基因组仍然会有很多确实区域,比如即便是最新的人类参考基因组,其中仍然会有超过百万的“N”。读长的提升能够有效提高基因组组装的完整性。
准确性:基因组组装的准确性可以在核酸水平或者结构变异水平进行描述。Illumina的三代测序技术的准确性非常高,每个碱基准确性>99.9%,PacBio和Nanopore的准确性在足够测序深度的情况下,经过算法校正之后也能够达到99.9%。对于PacBio测序而言,其准确性主要是受到随机的插入缺失突变的影响。而Nanopore的准确性会受到一些非随机因素的影响,比如共聚物序列,因而其准确性要落后于PacBio。在基因组结构水平上的准确性主要受重复序列的影响。重复序列可能会被认为是同一个序列区域。长读长测序能够减少这种错误,3.6kbp的读长与150kbp的读长相比,组装错误多了10倍。
总结
三代测序技术极大的提升了基因组的质量。虽然说20X的测序能够对一个基因组的组装已经足够了,但是还是建议>75X,这样有足够的测序深度能够对三代测序中的错误进行有效的校正。如果预算和样本允许,建议只对校正后深度>20X,长度>20kbp的测序片段进行组装。同时,测序技术发展十分迅速,在未来我们可以有更高质量的基因组,更低的花费。
==== THE END ===
参考资料:
Lee, H., Gurtowski, J., Yoo, S., Nattestad, M., Marcus, S., Goodwin, S., ... & Schatz, M. (2016). Third-generation sequencing and the future of genomics. BioRxiv, 048603.
Bellec, A., Courtial, A., Cauet, S., Rodde, N., & Vautrin, S. (2016). Long Read Sequencing Technology to Solve Complex Genomic Regions Assembly in Plants. Next Generat Sequenc & Applic, 3(128), 10-4172.


- 在Pi中寻找幸运数字
- Python助力2020高校应届生就业 获取2020校园招聘信息
- 第一次接触c语言
- Python助力2020考研调剂 各大高校调剂信息(持续更新)
- Linux中标准输出和标准错误的重导向
- Python助力2020高校应届生就业 获取2020校园招聘信息
- 雌性对雄性的选择是相关性状进化的驱动力
- Python助力2020考研调剂 各大高校调剂信息(持续更新)
- Python助力2020考研调剂 获取2020考研 高校调剂信息
- 进化重测序实验
- 7. JDK拍了拍你:字符串拼接一定记得用MessageFormat#format
- 项目实战:Qt中英文输入软键盘(支持Qt4、Qt5、触摸和键鼠混合输入等)
- LaTex零基础入门
- 7. JDK拍了拍你:字符串拼接一定记得用MessageFormat#format
- 5行Python代码调用电脑摄像头
- 爬取某婚恋网站数千小姐姐头像
- Python实现图片相似度计算
- 基于verilog实现数据检测-基于状态机的数据检测
- 支持 gRPC 长链接,深度解读 Nacos 2.0 架构设计及新模型
- PHP代码审计05之正则使用不当