生信小白学习日记-day1——NGS基础 FASTQ格式解释和质量评估
2019年5月25日,一个普通的周六,正在听的歌——北京东路的日子,开始学习生信,写博客。
说明:阅读生信宝典和查阅文章的总结,原文请关注公众号生信宝典,参考的博文都附有链接,仅供参考。
生信宝典
系列教程
关于编程学习的一些思考
知乎专栏:https://zhuanlan.zhihu.com/Data-Analysis
这篇文章讲述两个问题:系统学习还是遇到问题再找答案?是否要写博客。
- 第一个问题,两种途径都可以,都可以成为大神,但其一,要付出足够多的时间去写代码,修复bug;其二,要多思考、总结,在系统学习时给自己出题,在遇到问题时弄清楚一行行代码的意义;其三,要有兴趣和信仰,对代码的信仰、遇到bug时的信仰、资源的信仰、博客的信仰,相信自己,也相信资源。
- 第二个问题,参考第一个问题的其三。
这篇文章提到几个可能有用的书和文档:
- 廖雪峰老师的 python 教程
- R语言,dplyr data.table ggplot2 包的帮助文档
- stackoverflow 网站几乎能找到所有编程问题的答案
NGS基础——FASTQ格式解释和质量评估
一些复制粘贴,一些注释总结,仅助于自己记忆(记忆力相当差,把能记的都记了)和理解,当然可能有错误,后续再慢慢改正吧
关掉音乐专心学习
FASTQ文件格式和命名
- 用gzip压缩,一般我们都用双端测序,返回两个FASTQ文件,左端和右端分别命名为_1或R1, _2或R2。 如:sample_name_1_1.fq.gz,sample_name_1_2.fq.gz(第一个下划线后面的数字为重复,第二个下划线后面的数字指定哪一端)
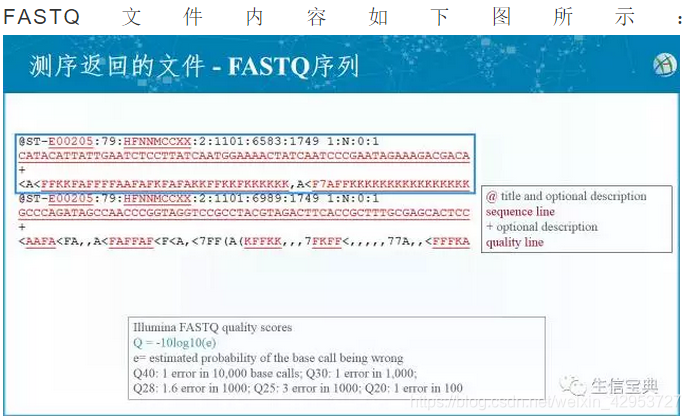
- 第一行以@开头,后面是reads 的ID 以及其他信息。
- 第二行为read序列。
- 第三行以+开头,一般后面无内容;若有则为序列名字,与第一行相同。
- 第四行为reads质量值。若该碱基测序出错率p_error 为0.001,则Q为30,换算公式为:Q = -10log(p_error)。而测序数据多采用Phred33编码,所以30+33=63,那么63对应的ASCII码为xx,一般碱基质量从0-40,因此ASCII码从(0+33)到(40+33)。以下表显示更为清楚:

- 为了检测传输的FASTQ文件是否完整,一般会附带一个MD5校验文件
MD5值
#获取MD5值 ct@ehbio:~/$ md5sum test.fq.gz | tee > MD5.txt # tee: 同时写入文件并在屏幕输出结果 + d41d8cd98f00b204e9800998ecf8427e test.fq.gz #输出文字 #验证MD5值 ct@ehbio:~/$ md5sum -c MD5.txt test.fq.gz: 确定 #输出文字
测序质量评估
测序质量可以从两个水平评估:测序reads数
和 测序碱基数
。它们都可以用简单的bash命令进行计算,但不是最高效的方式。

ct@ehbio:~/ $ zcat test.fq.gz #zcat: 不解压缩就可以查看文件内容 @ehbio1 ACCGAGCTTTTTTTTTTTTTT + ????????????????????? @ehbio2 ACCGAGCTTTTTTTTTTTTTT + ????????????????????? @ehbio3 ACCGAGCTTTTTTTTTTTTTT + ????????????????????? #获取行数,然后除以4,得到reads数 ct@ehbio:~/ $ echo $(( `zcat test.fq.gz | wc -l` /4 )) 4 #echo: 这里用于显示变量,还可用于-n,输出后不换行,-e,开启转义; #$(( )): 将其他进制转成十进制数显示出来; #``与$()都用来重组命令行,先执行``或()里的命令,将结果替换出来,再组成新的命令行。 #wc: 用于计算文件的字数、行数、列数等,-l 只显示行数。 #获取碱基数 ct@ehbio:~/ $ zcat test.fq.gz | grep -A 1 '^@' | grep '^[ACGTN]' | wc -c 66 #查看test.fq.gz内容,从中获取第一列开头为@的行及其之后的内容,从中获取开头为ACGTN的行,计算字符数。 #grep -A<显示列数>或--after-context=<显示列数> 除了显示符合范本样式的那一列之外,并显示该列之后的内容。
获得reads数和碱基数后,可以用表格展示。但为了更直观一般用柱状图展示。样品特别多时用直方图展示,优先关注是否存在测序量异常高或低的样品。
测序质量
一般用FastQC软件来评估测序质量,它是一个基于Java的软件包(并不清楚Java是啥),下载后放到环境变量就可使用。
- 运行方式:fastqc ehbio_1_1.fq.gz ehbio_1_2.fq.gz
- 生成结果和解释如表所示:


左图显示每个碱基的中位质量得分(箱线图中间的蓝线)都比较高,而右图的的碱基质量得分变化较大,测序质量逐渐下降,测序质量差。可能需要进行一定的预处理比如移除低质量碱基。

图横坐标表示平均GC含量,纵坐标表示reads数。左图显示每个read的GC分布(红线)与理论分布(蓝线)相契合,GC含量均一。右图出现了GC含量双峰,表示测序样品可能存在特定的序列污染如混入了引物二聚体,当这一指标异常并且导致后期的序列比对率很低时,需要引起注意。

接头序列含量图。横坐标表示read中碱基的位置,纵坐标表示接头序列含量。从图中可以得知read的前17 bp是干净的,之后的位置出现了比例不等的接头序列,需要去除。另一方面也反映了这个库的质量很差,有可能需要重建。
当样品数目很多时,会得到更多的质量评估图,结果很不直观。因此可以用下面所示的所有样品测序质量的散点图来表示。通过GC含量(横坐标)和碱基测序质量(纵坐标)来判断样品的测序质量。

两个值都是FAIL的样品,需要重点关注。
- 附grep命令详解:https://www.geek-share.com/detail/2664289520.html
贴来一些
1.主要参数
[options]主要参数:
-a或–text 不要忽略二进制的数据。
-A<显示列数>或–after-context=<显示列数> 除了显示符合范本样式的那一列之外,并显示该列之后的内容。
-b或–byte-offset 在显示符合范本样式的那一列之前,标示出该列第一个字符的位编号。
-B<显示列数>或–before-context=<显示列数> 除了显示符合范本样式的那一列之外,并显示该列之前的内容。
-c或–count 计算符合范本样式的列数。
-C<显示列数>或–context=<显示列数>或-<显示列数> 除了显示符合范本样式的那一列之外,并显示该列之前后的内容。
-d<进行动作>或–directories=<进行动作> 当指定要查找的是目录而非文件时,必须使用这项参数,否则grep指令将回报信息并停止动作。
-e<范本样式>或–regexp=<范本样式> 指定字符串做为查找文件内容的范本样式。
-E或–extended-regexp 将范本样式为延伸的普通表示法来使用。
-f<范本文件>或–file=<范本文件> 指定范本文件,其内容含有一个或多个范本样式,让grep查找符合范本条件的文件内容,格式为每列一个范本样式。
-F或–fixed-regexp 将范本样式视为固定字符串的列表。
-G或–basic-regexp 将范本样式视为普通的表示法来使用。
-h或–no-filename 在显示符合范本样式的那一列之前,不标示该列所属的文件名称。
-H或–with-filename 在显示符合范本样式的那一列之前,表示该列所属的文件名称。
-i或–ignore-case 忽略字符大小写的差别。
-l或–file-with-matches 列出文件内容符合指定的范本样式的文件名称。
-L或–files-without-match 列出文件内容不符合指定的范本样式的文件名称。
-n或–line-number 在显示符合范本样式的那一列之前,标示出该列的列数编号。
-q或–quiet或–silent 不显示任何信息。
-r或–recursive 此参数的效果和指定“-d recurse”参数相同。
-s或–no-messages 不显示错误信息。
-v或–revert-match 反转查找。
-V或–version 显示版本信息。
-w或–word-regexp 只显示全字符合的列。
-x或–line-regexp 只显示全列符合的列。
-y 此参数的效果和指定“-i”参数相同。
–help 在线帮助。
pattern正则表达式主要参数:
\: 忽略正则表达式中特殊字符的原有含义。
^:匹配正则表达式的开始行。
$: 匹配正则表达式的结束行。
<:从匹配正则表达 式的行开始。
>:到匹配正则表达式的行结束。
[ ]:单个字符,如[A]即A符合要求 。
[ - ]:范围,如[A-Z],即A、B、C一直到Z都符合要求 。
. :所有的单个字符。
*: 所有字符,长度可以为0。
4.grep命令使用简单实例
$ grep ‘test’ d*
显示所有以d开头的文件中包含 test的行。
$ grep ‘test’ aa bb cc
显示在aa,bb,cc文件中匹配test的行。
$ grep ‘[a-z]{5}’ aa
显示所有包含每个字符串至少有5个连续小写字符的字符串的行。
$ grep ‘w(es)t.\1′ aa
如果west被匹配,则es就被存储到内存中,并标记为1,然后搜索任意个字符(.),这些字符后面紧跟着 另外一个es(\1),找到就显示该行。如果用egrep或grep -E,就不用”\”号进行转义,直接写成’w(es)t.*\1′就可以了。
5.grep命令使用复杂实例
假设您正在’/usr/src/Linux/Doc’目录下搜索带字符 串’magic’的文件:
$ grep magic /usr/src/Linux/Doc/*
sysrq.txt:* How do I enable the magic SysRQ key?
sysrq.txt:* How do I use the magic SysRQ key?
其中文件’sysrp.txt’包含该字符串,讨论的是 SysRQ 的功能。
默认情况下,’grep’只搜索当前目录。如果 此目录下有许多子目录,’grep’会以如下形式列出:
grep: sound: Is a directory
这可能会使’grep’ 的输出难于阅读。这里有两种解决的办法:
明确要求搜索子目录:grep -r
或忽略子目录:grep -d skip
如果有很多 输出时,您可以通过管道将其转到’less’上阅读:
$ grep magic /usr/src/Linux/Documentation/* | less
这样,您就可以更方便地阅读。
有一点要注意,您必需提供一个文件过滤方式(搜索全部文件的话用 *)。如果您忘了,’grep’会一直等着,直到该程序被中断。如果您遇到了这样的情况,按 ,然后再试。
下面还有一些有意思的命令行参数:
grep -i pattern files :不区分大小写地搜索。默认情况区分大小写,
grep -l pattern files :只列出匹配的文件名,
grep -L pattern files :列出不匹配的文件名,
grep -w pattern files :只匹配整个单词,而不是字符串的一部分(如匹配’magic’,而不是’magical’),
grep -C number pattern files :匹配的上下文分别显示[number]行,
grep pattern1 | pattern2 files :显示匹配 pattern1 或 pattern2 的行,
grep pattern1 files | grep pattern2 :显示既匹配 pattern1 又匹配 pattern2 的行。
grep -n pattern files 即可显示行号信息
grep -c pattern files 即可查找总行数
这里还有些用于搜索的特殊符号:
< 和 > 分别标注单词的开始与结尾。
例如:
grep man * 会匹配 ‘Batman’、’manic’、’man’等,
grep ‘<man’ * 匹配’manic’和’man’,但不是’Batman’,
grep ‘<man>’ 只匹配’man’,而不是’Batman’或’manic’等其他的字符串。
‘^’:指匹配的字符串在行首,
‘$’:指匹配的字符串在行 尾,
突然感到心累,grep命令好复杂T_T
今天就到这吧,后背酸痛,明天继续0.0

- 传智播客--WPF基础视频学习--sender解释(小白内容)
- 生信小白学习日记Day6——NGS分析流程(before bwa)
- zhu的学习日记:shell脚本学习一(基础)
- Java学习日记番外篇:jdbc基础
- 小白学习Machine Learning in Action-机器学习实战------Python基础
- 零基础如何开始学习 Python?看完这篇小白变小牛!
- linux 学习日记基础(转)
- Java基础第二十天学习日记
- # 运维小白的成长日记第五天-# 基础网络构建OSI七层模型-数据链路层基础知识
- Linux学习日记--基础命令(3)-文件操作,通配符,命令别名
- C++基础学习DAY1-02 双冒号作用域
- Unity3D小白学习日记(02):U3D如何连接SQL SERVER数据库(亲测可行!)
- Java学习日记12:xml基础
- 黑马程序员_学习日记43_609基础加强(多播委托、事件、程序集、反射)
- 基于c#的windows基础设计(学习日记1)【关于异或运算】
- Java零基础学习日记-配置jdk环境
- 黑马程序员Java学习日记(9)基础加强
- Linux学习日记 —— 10.5.1 Shell基础-Bash变量-数值运算与运算符
- 【DAY1】hadoop的安装配置基础学习笔记
- python学习日记2--基础语法篇