BWA-MEM原始论文算法部分翻译
2017-11-23 00:00
375 查看
Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM
2.1 Aligning a single query sequence 单序列比对
2.1.1 Seeding and re-seeding
BWA-MEM follows the canonical seed-and-extend paradigm. It initially seeds an alignment with supermaximal exact matches (SMEMs) using an algorithm we found previously (Li, 2012,Algorithm 5), which essentially finds at each query position the longest exact match covering the position. However, occasionally the true alignment may not contain any SMEMs. To reduce mismappings caused by missing seeds, we introduce re-seeding. Suppose we have a SMEM of length l with k
occurrences in the reference genome. If l is too large (over 28bp by default), we re-seed with the longest exact matches that cover the middle base of the SMEM and occur at least k + 1 times in the genome. Such seeds can be found by requiring a minimum occurrence in the original SMEM algorithm.
BWA-MEM算法基于seed-and-extend。首先使用以前的算法,用 supermaximal exact matches (SMEMs)来seeds一个比对,找到每个查询位点的 longest exact match。
(SMEMs: Formally, a maximal exact match (MEM)最大精确匹配 is a an exact match that cannot be extended in either direction of the match. An SMEM is a MEM that is not contained in other MEMs on the query sequence. https://academic.oup.com/bioinformatics/article/28/14/1838/218887)
然而,有时候真实的比对不一定包含任何SMEMs,为了减少丢失seeds引起的错误匹配,我们引入了re-seeding。假设有个SMEM,长度为l,在标准基因组中出现了k次。如果SMEM过长(超过28bp),我们对覆盖了SMEM中间碱基且至少出现k+1次的longest exact matches进行re-seed。这样的seeds可以通过设定最小出现次数在原始的SMEM算法中找到。
2.1.2 Chaining and chain filtering
We call a group of seeds that are colinear and close to each other as a chain. We greedily chain the seeds while seeding and then filter out short chains that are largely contained in a long chain and are much worse than the long chain (by default, both 50% and 38bp shorter than the long chain). Chain filtering aims to reduce unsuccessful seed extension at a later step. Each chain may not always correspond to a final hit. Chains detected here do not need to be accurate.
我们把一组线性或相近的seeds称作chain。seeding过程中尽可能将chain变长,同时过滤掉过短的chains,这些chains通常大量包含在长chain中。过滤chain的目的是为了减少下一步中不成功的seed extension。每个chain并不总是有最终的hit,这时候的chains不需要非常精确(?)
2.13 Seed extension
We rank a seed by the length of the chain it belongs to and then by the seed length. For each seed in the ranked list, from the best to the worst, we drop the seed if it is already contained in an alignment found before, or extend the seed with a banded affine-gap-penalty dynamic programming (DP) if it potentially leads to a new alignment.
我们依据seed所从属的chain的长度和seed的长度对seed进行排序。对于每一个在排好序的list中的seed,舍弃掉已经比对过的seed,或如果该seed可能产生一个新的alignment,利用某算法(a banded affine-gap-penalty dynamic programming (DP))对其进行extend。??
BWA-MEM’s seed extension differs from the standard seed extension in two aspects.
BWA-MEM的seed extension同标准的seed extension不同。
Firstly, suppose at a certain extension step we come to reference position x with the best extension score achieved at query position y. BWAMEM will stop extension if the difference between the best extension score and the score at (x, y) is larger than Z+|x−y|×pgapExt, where pgapExt is the gap extension penalty and Z is an arbitrary cutoff. This heuristic avoids extension through a poorly aligned region with good flanking alignment. It is similar to the X-dropoff heuristic in BLAST (Altschul et al., 1997), but does not penalize long gaps in one of the reference or the query sequences.
首先,设想一次可能的extension步骤,在标准基因组的位置(x,y)时达到最大扩展分数,如果最大扩展分数和(x,y)位置的分数差距大于Z+|x−y|×pgapExt时,BWAMEM停止扩展。pgapExt是gap 扩展扣分,Z是一个任意截断(?)。这种扩展式搜索使用旁侧比对可以回避掉不好的比对区域。这种方法同BLAST的X-dropoff扩展式搜索类似,但是在标准序列和比对序列上出现长gaps时不进行扣分。
Secondly, while extending a seed, BWA-MEM tries to keep track of the best extension score reaching the end of the query sequence. If the difference between the best score reaching the end and the best local alignment score is below a threshold, the local alignment will be rejected even if it has a higher score. BWA-MEMuses this strategy to automatically choose between local and end-to-end alignments. It may align through true variations towards the end of a read and thus reduces reference bias, while avoids introducing excessive mismatches and gaps which may happen when we force a chimeric read into an end-to-end alignment.
其次,扩展seed时,BWA-MEM记录到达序列末端的最大扩展得分,如果末端最大得分和局部比对得分之间的差距低于阈值时,将局部比对舍弃,即便局部得分更高。BWA-MEM使用这个策略来自动选择使用局部比对还是末端比对。算法略过真实的变异朝read末端比对,减少标准偏差,同时避免带来更多错误和gaps,这些错误和gaps发生在我们强制一个随机read执行端对端比对时。
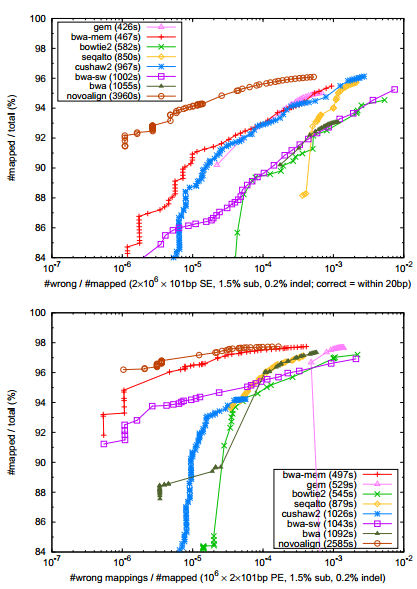
Fig. 1. Percent mapped reads as a function of the false alignment rate under different mapping quality cutoff. Alignments with mapping quality 3 or lower are excluded. An alignment is wrong if after correcting clipping, its start position is within 20bp from the simulated position. 106 pairs of
101bp reads are simulated from the human reference genome using wgsim (http://bit.ly/wgsim2) with 1.5% substitution errors and 0.2% indel variants. The insert size follows a normal distribution N(500, 502). The reads are aligned back to the genome either as single end (SE; top panel) or as paired end (PE; bottom panel). GEM is configured to allow up to 5 gaps and to output suboptimal alignments (option ‘–e5 –m5 –s1’ for SE and ‘–e5 –m5 –s1 –pb’ for PE). GEM does not compute mapping quality. Its mapping quality is estimated with a BWA-like algorithm with suboptimal alignments available. Other mappers are run with the default setting except for specifying the insert size distribution. The run time in seconds on a single CPU core is shown in the parentheses.
2.2 Paired-end mapping双端映射
2.2.1 Rescuing missing hits 恢复丢失的hits
Like BWA (Li and Durbin, 2009), BWAMEM processes a batch of reads at a time. For each batch, it estimates the mean µ and the variance σ2 of the insert size distribution from reliable single-end hits. For the top 100 hits (by default) of either end, if the mate is unmapped in a window [µ - 4σ, µ + 4σ] from each hit, BWA-MEM performs SSE2-based Smith-Waterman alignment (SW; Farrar 2007) for the mate within the window. The second best SW score is recorded to detect potential mismapping in a long tandem repeat. Hits found from both the single-sequence alignment and SW rescuing will be used for pairing.
同BWA类似,BWA-MEM一次处理一包reads。
2.2.2 Pairing
Given the i-th hit for the first read and j-th hit for the second, BWA-MEM computes their distance dij if the two hits are in the right orientation, or sets dij to infinity otherwise. It scores the pair (i, j)
with Sij = Si + Sj - min{-a log4 P (dij), U}, where Si and Sj are the SW score of the two hits, respectively, a is the matching score and P (d) gives the probability of observing an insert size larger than d assuming a normal distribution. ‘log4’ arises when we interpret SW score as odds
ratio (Durbin et al., 1998). U is a threshold that controls pairing: if dijis small enough such that -a log4 P (dij) < U, BWA-MEM prefers to pair the two ends; otherwise it prefers the unpaired alignments. BWA-MEM chooses the pair (i, j) that maximizes Sij as the final alignments for both
ends. This pairing strategy jointly considers the alignment scores, the insert size and the possibility of chimeric read pairs.
2.1 Aligning a single query sequence 单序列比对
2.1.1 Seeding and re-seeding
BWA-MEM follows the canonical seed-and-extend paradigm. It initially seeds an alignment with supermaximal exact matches (SMEMs) using an algorithm we found previously (Li, 2012,Algorithm 5), which essentially finds at each query position the longest exact match covering the position. However, occasionally the true alignment may not contain any SMEMs. To reduce mismappings caused by missing seeds, we introduce re-seeding. Suppose we have a SMEM of length l with k
occurrences in the reference genome. If l is too large (over 28bp by default), we re-seed with the longest exact matches that cover the middle base of the SMEM and occur at least k + 1 times in the genome. Such seeds can be found by requiring a minimum occurrence in the original SMEM algorithm.
BWA-MEM算法基于seed-and-extend。首先使用以前的算法,用 supermaximal exact matches (SMEMs)来seeds一个比对,找到每个查询位点的 longest exact match。
(SMEMs: Formally, a maximal exact match (MEM)最大精确匹配 is a an exact match that cannot be extended in either direction of the match. An SMEM is a MEM that is not contained in other MEMs on the query sequence. https://academic.oup.com/bioinformatics/article/28/14/1838/218887)
然而,有时候真实的比对不一定包含任何SMEMs,为了减少丢失seeds引起的错误匹配,我们引入了re-seeding。假设有个SMEM,长度为l,在标准基因组中出现了k次。如果SMEM过长(超过28bp),我们对覆盖了SMEM中间碱基且至少出现k+1次的longest exact matches进行re-seed。这样的seeds可以通过设定最小出现次数在原始的SMEM算法中找到。
2.1.2 Chaining and chain filtering
We call a group of seeds that are colinear and close to each other as a chain. We greedily chain the seeds while seeding and then filter out short chains that are largely contained in a long chain and are much worse than the long chain (by default, both 50% and 38bp shorter than the long chain). Chain filtering aims to reduce unsuccessful seed extension at a later step. Each chain may not always correspond to a final hit. Chains detected here do not need to be accurate.
我们把一组线性或相近的seeds称作chain。seeding过程中尽可能将chain变长,同时过滤掉过短的chains,这些chains通常大量包含在长chain中。过滤chain的目的是为了减少下一步中不成功的seed extension。每个chain并不总是有最终的hit,这时候的chains不需要非常精确(?)
2.13 Seed extension
We rank a seed by the length of the chain it belongs to and then by the seed length. For each seed in the ranked list, from the best to the worst, we drop the seed if it is already contained in an alignment found before, or extend the seed with a banded affine-gap-penalty dynamic programming (DP) if it potentially leads to a new alignment.
我们依据seed所从属的chain的长度和seed的长度对seed进行排序。对于每一个在排好序的list中的seed,舍弃掉已经比对过的seed,或如果该seed可能产生一个新的alignment,利用某算法(a banded affine-gap-penalty dynamic programming (DP))对其进行extend。??
BWA-MEM’s seed extension differs from the standard seed extension in two aspects.
BWA-MEM的seed extension同标准的seed extension不同。
Firstly, suppose at a certain extension step we come to reference position x with the best extension score achieved at query position y. BWAMEM will stop extension if the difference between the best extension score and the score at (x, y) is larger than Z+|x−y|×pgapExt, where pgapExt is the gap extension penalty and Z is an arbitrary cutoff. This heuristic avoids extension through a poorly aligned region with good flanking alignment. It is similar to the X-dropoff heuristic in BLAST (Altschul et al., 1997), but does not penalize long gaps in one of the reference or the query sequences.
首先,设想一次可能的extension步骤,在标准基因组的位置(x,y)时达到最大扩展分数,如果最大扩展分数和(x,y)位置的分数差距大于Z+|x−y|×pgapExt时,BWAMEM停止扩展。pgapExt是gap 扩展扣分,Z是一个任意截断(?)。这种扩展式搜索使用旁侧比对可以回避掉不好的比对区域。这种方法同BLAST的X-dropoff扩展式搜索类似,但是在标准序列和比对序列上出现长gaps时不进行扣分。
Secondly, while extending a seed, BWA-MEM tries to keep track of the best extension score reaching the end of the query sequence. If the difference between the best score reaching the end and the best local alignment score is below a threshold, the local alignment will be rejected even if it has a higher score. BWA-MEMuses this strategy to automatically choose between local and end-to-end alignments. It may align through true variations towards the end of a read and thus reduces reference bias, while avoids introducing excessive mismatches and gaps which may happen when we force a chimeric read into an end-to-end alignment.
其次,扩展seed时,BWA-MEM记录到达序列末端的最大扩展得分,如果末端最大得分和局部比对得分之间的差距低于阈值时,将局部比对舍弃,即便局部得分更高。BWA-MEM使用这个策略来自动选择使用局部比对还是末端比对。算法略过真实的变异朝read末端比对,减少标准偏差,同时避免带来更多错误和gaps,这些错误和gaps发生在我们强制一个随机read执行端对端比对时。
Fig. 1. Percent mapped reads as a function of the false alignment rate under different mapping quality cutoff. Alignments with mapping quality 3 or lower are excluded. An alignment is wrong if after correcting clipping, its start position is within 20bp from the simulated position. 106 pairs of
101bp reads are simulated from the human reference genome using wgsim (http://bit.ly/wgsim2) with 1.5% substitution errors and 0.2% indel variants. The insert size follows a normal distribution N(500, 502). The reads are aligned back to the genome either as single end (SE; top panel) or as paired end (PE; bottom panel). GEM is configured to allow up to 5 gaps and to output suboptimal alignments (option ‘–e5 –m5 –s1’ for SE and ‘–e5 –m5 –s1 –pb’ for PE). GEM does not compute mapping quality. Its mapping quality is estimated with a BWA-like algorithm with suboptimal alignments available. Other mappers are run with the default setting except for specifying the insert size distribution. The run time in seconds on a single CPU core is shown in the parentheses.
2.2 Paired-end mapping双端映射
2.2.1 Rescuing missing hits 恢复丢失的hits
Like BWA (Li and Durbin, 2009), BWAMEM processes a batch of reads at a time. For each batch, it estimates the mean µ and the variance σ2 of the insert size distribution from reliable single-end hits. For the top 100 hits (by default) of either end, if the mate is unmapped in a window [µ - 4σ, µ + 4σ] from each hit, BWA-MEM performs SSE2-based Smith-Waterman alignment (SW; Farrar 2007) for the mate within the window. The second best SW score is recorded to detect potential mismapping in a long tandem repeat. Hits found from both the single-sequence alignment and SW rescuing will be used for pairing.
同BWA类似,BWA-MEM一次处理一包reads。
2.2.2 Pairing
Given the i-th hit for the first read and j-th hit for the second, BWA-MEM computes their distance dij if the two hits are in the right orientation, or sets dij to infinity otherwise. It scores the pair (i, j)
with Sij = Si + Sj - min{-a log4 P (dij), U}, where Si and Sj are the SW score of the two hits, respectively, a is the matching score and P (d) gives the probability of observing an insert size larger than d assuming a normal distribution. ‘log4’ arises when we interpret SW score as odds
ratio (Durbin et al., 1998). U is a threshold that controls pairing: if dijis small enough such that -a log4 P (dij) < U, BWA-MEM prefers to pair the two ends; otherwise it prefers the unpaired alignments. BWA-MEM chooses the pair (i, j) that maximizes Sij as the final alignments for both
ends. This pairing strategy jointly considers the alignment scores, the insert size and the possibility of chimeric read pairs.

相关文章推荐
- 扫描线算法的论文《The Intersections for a Set of 2D Segments, and Testing Simple Polygons》的部分翻译
- Dremel: Interactive Analysis of Web-Scale Datasets 1~6节算法思想部分翻译
- 分布式一致性算法:Raft 算法(论文翻译)
- 砥志研思SVM(四) 序列最小最优化算法(SMO)论文翻译
- Raft算法官方论文中文翻译
- CVPR 2017论文集锦(论文分类)—— 附录部分翻译
- 路由模拟——论文算法设计部分(2)
- 分布式一致性算法:Raft 算法(Raft 论文翻译)
- 路由模拟——论文算法设计部分(3)
- BWA-MEM算法记录
- Raft一致性算法论文的中文翻译
- Raft算法国际论文全翻译
- 路由模拟——论文算法设计部分(1)
- 路由模拟——论文算法设计部分(4)
- Borg论文翻译 以及部分理解
- [Suzuki85]轮廓跟踪算法论文翻译
- 替罪羊树 论文部分翻译
- Traking-Learning-Detection TLD经典论文部分翻译
- 分布式一致性算法:Raft 算法(论文翻译)
- [Suzuki85]轮廓跟踪算法论文翻译